
医療機器及び体外診断用医薬品の製造管理及び品質管理の基準(QMS)について
医療機器及び体外診断用医薬品の製造管理及び品質管理の基準は、医療機器等を製造販売する際の承認(認証)要件として制定された厚生労働省令です。
なお、本基準はISO13485に準拠する内容で制定されました。
平成16年12月17日厚生労働省令第169号
薬事法(昭和35年法律第145号)第14条第2項第4号及び第19条の2第5項において準用する第14条第2項第4号の規定に基づき、医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令を次のように定める。
医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令
目次
第一章総則(第1条-第3条)
第二章医療機器製造業者等の製造所における製造管理及び品質管理
第一節通則(第4条)
第二節品質管理監督システム(第5条-第9条)
第三節管理監督者の責任(第10条-第20条)
第四節資源の管理監督(第21条-第25条)
第五節製品実現(第26条-第53条)
第六節測定、分析及び改善(第54条-第64条)
第三章医療機器保管等製造業者等の製造所における製造管理及び品質管理(第65条-第72条)
第四章生物由来医療機器等製造業者等の製造所における製造管理及び品質管理(第73条-第79条)
第五章体外診断用医薬品製造業者等の製造所における製造管理及び品質管理(第80条)
附則
ここでは、QMS省令の中で第二章医療機器製造業者等の製造所における製造管理及び品質管理において、どのようなことが求められているか、基本的な考え方のみを説明します。具体的な内容については、QMS省令、施行通知、関係通知等によりご確認ください。
QMS省令に定められた要求事項について
品質管理監督システム(第5条-第9条)
第5条では品質管理監督システムに係る要求事項が定められています。
品質管理監督システムは第2条(定義)において「製造業者及び外国製造業者が品質に関して製造所の管理監督を行うためのシステムをいう。」と定められています。
QMS省令ではこれまでの製造を中心とした品質管理の基準であるGMPから企業全体の品質活動である品質管理監督システムを構築することが求められています。このため、品質管理監督システムの対象は製造部門だけではなく、技術、工場、営業等全部門が対象となります。
製造業者等は品質管理監督システムを確立し、実施し、実効性を維持しなければなりません。具体的には次の業務を行うべきとされています。
-
- 品質管理監督システムに必要な工程(プロセス)の範囲を確定します(どの工程を管理するべきか、工程の順序や関連性を明確にします。)。
- 工程の判定基準や判定方法を明確にします。
- 工程の実施や監視測定に必要な体制を確保します。
- 工程を監視測定、分析し、その結果により必要な是正措置、予防措置につなげていきます。
- 実効性を維持します。→PDCAサイクルによって、維持していきます。
第6条-第9条では品質管理監督システムに係る文書類に関することが定められています。
製造業者等は品質管理監督システムに必要な文書を作成し、その規定に従って実行し、記録を残さなければなりません。
主な文書類を次にあげます。
-
- 品質方針表明書、品質目標表明書
- 品質管理監督システム基準書(QMS省令の要求事項に従い、品質管理監督システムに係るすべての業務を網羅した最高位の品質管理規定です。ISOでいう品質マニュアルと同様です。)
- 製造所の工程について、計画的な実施や管理がなされるようにするために必要な文書類
品質管理監督システムに必要な文書類は、その妥当性を照査し、内容の更新、変更内容の識別等適切な文書管理体制の下に管理されなければなりません。
また、文書の保管期間についても定められています。
- 特定保守管理医療機器に係る文書:15年間or有効期間+1年間の長い期間
- 特定保守管理医療機器以外に係る文書:5年間or有効期間+1年間の長い期間
管理監督者の責任(第10条-第20条)
第10条-第20条では管理監督者に係る要求事項が定められています。
管理監督者は第2条(定義)において「業務を行う役員等製造所の管理監督を行う者」と定められています。管理監督者は品質管理監督システムの運用に責任をもって関与することが求められています。
主な要求事項は次のとおりです。
- 適切な品質方針を定め、製造所において周知、理解させなければなりません。また、製造所の関係部門において、品質目標を定められるようにする必要があります。
- 製品受領者(製造販売業者等)の要求事項に応えるようにしなければなりません。この要求事項はGQPによる取決め等により明確にされていなければなりません。
- 責任技術者に対し、工程が確立、実施され、実効性が維持されているように責任及び権限を与えなければなりません。
- 管理監督者照査(マネジレントレビュー)を品質管理監督システムの計画通り実施しなければなりません。この照査では次に示す入力情報を照査し、出力情報を得て、所要の措置をとる必要があります。
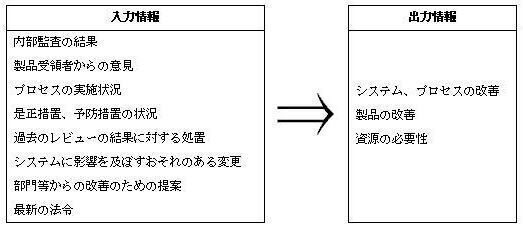
資源の管理監督(第21条-第25条)
資源とは第2条(定義)において「個人の有する知識及び技能並びに技術、設備その他の製造所の業務に活用される資源」と定められています。
第22条-第23条では人的な資源に関する要求事項が定められています。
職員は所要の技能及び経験を有し、かつ適切な教育訓練を受けていることが求められています。また、教育訓練は手順書に基づき実施されその記録を作成します。
第24条-第25条では物的な資源に関する要求事項が定められています。
業務運営基盤は「製造所における業務に必要な施設、設備及びサービスの体系」であり、製品要求事項への適合の達成に必要な業務運営基盤を適切に維持する必要があります。この中には作業所や製造設備だけでなく、輸送や情報伝達等製造を支援するサービスを含みます。
また、次に挙げる品目については薬局等構造設備規則に上乗せで求められる設備があります。
- 防じん、防湿、防虫、防その必要な製品
- 有毒ガスを取り扱う製品
- 液体状、ゾル状、ゲル状、粉末状の製品
作業環境についても、必要な作業環境を明確にし、管理する必要があります。職員との接触等により製品の品質に悪影響を及ぼすおそれがある場合には職員の健康状態や作業衣等に係る要求事項書を作成し、その手順に従い管理がされるようにしなければなりません。
製品実現(第26条-第53条)
第26条では製品実現計画についての要求事項が定められています。
製品実現に必要な工程について計画を確立します。この計画では、製品実現に係る工程以外の工程との整合性をとったものである必要があります。計画の策定には次の事項を明確にします。
- 製品の品質目標、製品要求事項
- 製品に特有のプロセス、文書、資源
- 製品固有のバリデーション等、製造出荷可否決定基準
- 必要な記録類
また、製品実現に係る全ての工程におけるリスクマネジメントに係る要求事項書や記録の作成も求められています。
第27条-第29条では製品要求事項に関する要求事項が定められています。
製品受領者(製造販売業等)の要求事項など、製品に求められる事項を明確にします。この内容については、法令の規定や自社で定める要求事項も含まれます。これらの製品要求事項は適切に照査されていなければなりません。また、製品受領者との情報伝達の手法についても適切に定めておく必要があります。
第30条-第36条では設計開発に関する要求事項が定められています。
製品の設計開発のための手順書を作成し、設計開発計画を策定する必要があります。設計開発計画は、設計開発の各段階それぞれにおいて適切な照査、検証等が必要となりこれらを明確にします。
設計開発における入力情報と出力情報は次のとおり定められています。
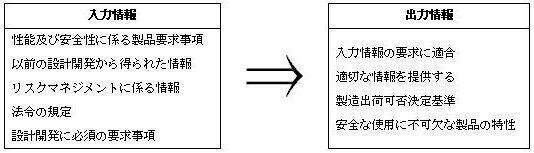
設計開発について、その適切な段階において、体系的な照査、検証、バリデーションを実施する必要があります。また、変更時にはその内容について手順に従い適切な管理の下に変更を行います。
第37条-第39条では購買工程に関する要求事項が定められています。
購買工程では、購買物品要求事項に適合するようにするための手順書を作成します。購買物品の供給者の選定については、自社で定めた判定基準に基づいて評価し、その記録を残す必要があります。また、購買物品の出荷可否決定、供給者側の手順、供給者の職員の適格性の確認等の購買情報を定め、購買物品の検証についても求められています。
第40条では製造及びサービス提供に関する要求事項が定められています。
製造所における製造及びサービス提供について、計画を策定し、次の管理条件の下で実施する必要があります。
- 製品の特性を記述した情報が利用できる体制である
- 手順書、要求事項書、作業指図書等が利用できる体制である
- 製造に見合う設備、器具を使用する
- 監視測定のための設備、器具が利用できる体制である
- 監視測定を実施する
- 工程の完了の許可、製造所出荷可否決定、出荷及び出荷業務を適切に実施する
- 手順書等に定められた包装及び表示に係る作業を実施する
第44条、第46条では滅菌製品に関する要求事項が定められています。
滅菌製品については、滅菌工程の指標について管理が求められます。また、業務運営基盤について、次の上乗せ基準があります。
- 製造工程に応じ、じんあい又は微生物汚染防止するのに必要な構造設備
- 作業管理区域の清浄度の確保
- 製造工程に応じ、必要な質と量の製造用水供給設備
- 必要な滅菌装置
- 滅菌工程の管理に必要な設備器具
第45条ではバリデーションに関する要求事項が定められています。
製造工程等のバリデーションは、製造及びサービス提供に係る工程について、それ以降の監視測定でその工程の結果の検証ができない場合、実施する必要があります。バリデーション対象工程については、次の事項に係る実施要領を定めます。
- 当該工程の照査等のための判定基準
- 設備、器具、職員の適格性
- 方法及び手順
- 再バリデーション
ソフトウェアの適用についても、手順書に従いバリデーションを行う必要があります。
第47条-第50条では製品等の識別や追跡可能性に関する要求事項が定められています。
製品実現に係る全ての工程において、製品の識別表示を行う必要があります。特に不適合品との区分を確実に行うよう、識別表示による区分に係る手順書を作成しなければなりません。また、製品の状態の識別表示による区分は、製品の製造、保管のみならず、設置及び付帯サービス業務に係るすべての工程において維持しなければなりません。
追跡可能性とは、第2条(定義)において「履歴、適用又は所在を追跡できる状態にあること」と定められています。追跡可能性の確保に係る手順書を作成し、製品ごとに追跡可能性の確保の程度やそのために必要な記録について定める必要があります。
特定医療機器について、構成部品等又は作業環境の条件によって製品に影響がある恐れがある場合には、これら条件の全てに係る記録の追跡可能性を確保しなければなりません。また、特定医療機器に係る製品の荷受人の氏名、住所を記録されるようにします。
第53条では監視測定のための設備及び器具の管理に関する要求事項が定められています。
製品要求事項への適合性を実証するために必要な監視測定を明確にし、また、このために必要な設備及び器具についても明確に規定します。これらの設備及び器具については、適切な校正、調整がなされており、それらの状態が明確に識別表示されている必要があります。
これらの設備及び器具の不適合が判明した場合には、これらで判定を行った従前の監視測定結果について、その妥当性を評価し、適切な措置をとらなければなりません。
測定、分析、改善(第54条-第64条)
第54条-第59条では監視測定についての要求事項が定められています。
製品の適合性を実証するために、監視測定を行う工程を明確とし、監視測定の方法、基準等について計画を策定しなければなりません。製品が製品要求事項に適合していることを検証するために、工程の適切な段階においてそれぞれ必要な監視測定を実施し、その結果を記録することが求められています。
また、品質管理監督システムの実施状況について、あらかじめ定めた間隔で内部監査を実施しなければなりません。内部監査における主な要求事項は次のとおりです。
- 内部監査の対象工程の重要性や従前の監査結果を考慮して、内部監査実施計画を策定しなければなりません。
- 内部監査の判定基準、範囲、頻度、方法を定めなければなりません。
- 内部監査員の確保し、内部監査員には自らの業務を内部監査させてはいけません。
- 内部監査実施計画の策定、実施、結果の報告、結果の記録について手順書で定めなければなりません。
- 発見された不適合及びその原因の除去を速やかに、確実に行い、その措置の検証を行わなければなりません。
第60条では不適合製品の管理についての要求事項が定められています。
製品要求事項に適合しない不適合製品については、意図に反した使用、操作や出荷がなされないよう識別し、区分がなされていなければなりません。不適合製品については、その処理に係る管理、それに関連する責任や権限を手順書に定める必要があります。
不適合製品は次のいずれかの方法によって処理します。
- 発見された不適合を除去するための措置をとる。
- 特別採用の下で、使用や操作の許可、次工程への許可、製造所からの出荷の決定を行う。
- 本来の意図された使用、操作、適用ができないようにするための措置をとる。
ここで、不適合に対して取られた措置についてはその記録を残し、特別採用を行った場合は、それを許可した職員を特定する記録を残す必要があります。また、不適合製品に修正を行った場合、その修正後の製品への再検証も必要になります。
第61条ではデータの分析についての要求事項が定められています。
品質管理監督システムが適切かつ実効性のあるものであることを実証するために、適切なデータを明確にし、収集、分析するための手順書を作成しなければなりません。
このデータ分析では、分析のみでなく解析、判断までを行う必要があります。
第62条では改善についての要求事項が定められています。
内部監査、データ分析、是正措置、予防措置、管理監督者照査を活用し、品質管理監督システムの妥当性や実効性を維持するために、随時改善を図らなければなりません。また、製品受領者の苦情への対応についても、適切に対応しその結果を記録することが求められています。
第63条では是正措置についての要求事項が定められています。
発見された不適合による影響に応じて、適切な是正措置をとらなければなりません。是正措置についてはフロー及び判断基準を明確にしておく必要があるため、次の事項を満たした手順書を作成する必要があります。
- 不適合の照査
- 原因の明確化
- 再発防止措置の適格性
- 是正措置の明確化及び実施
- 結果の記録
- とった是正措置及び実効性についての照査
第64条では予防措置についての要求事項が定められています。
起こりうる問題の影響に応じて、適切な予防措置をとらなければなりません。予防措置が必要とされる度合いを数値化し、順番に対処することが合理的であると考えられます。次の事項を満たした手順書を作成する必要があります。
- 起こり得る不適合とその原因の明確化
- 予防措置の必要性の評価
- 予防措置の明確化及び実施
- 結果の記録
- とった予防措置及び実効性についての照査
以上、QMS省令第二章において要求されている内容の主な点について記述しましたが、この他、生物由来医療機器等の製造所における製造管理及び品質管理として上乗せされた基準(第四章)が規定されています。また、保管等製造所については別途規定(第三章)されていますので、ご注意ください。
このページに関するお問い合わせ
健康福祉部生活衛生局薬事課
〒420-8601 静岡市葵区追手町9-6
電話番号:054-221-2869
ファクス番号:054-221-2199
yakuji@pref.shizuoka.lg.jp